Дисплазия соединительной ткани
Дисплазия соединительной ткани (от др.-греч. δυσ- — приставка, отрицающая положительный смысл слова и πλάσις — «образование, формирование») — генетически детерминированные состояния, характеризующиеся дефектами волокнистых структур и основного вещества соединительной ткани, приводящие к нарушению формообразования органов и систем, имеющие прогредиентное течение. Синонимы: соединительнотканная дисплазия, врождённая соединительнотканная недостаточность, наследственная коллагенопатия[3].
| Гипермобильный синдром | |
|---|---|
 | |
| МКБ-10 | M35.7 |
| МКБ-10-КМ | M35.7 |
| МКБ-9 | 728.5 |
| МКБ-9-КМ | 728.5[1][2] |
| OMIM | 147900 |
| DiseasesDB | 31101 |
| MedlinePlus | 003295 |
| MeSH | D007593 |
Выделяют дифференцированную и недифференцированную дисплазию соединительной ткани (ДСТ). Дифференцированная ДСТ включает в себя синдромы Элерса-Данлоса, Марфана, Стиклера, несовершенного остеогенеза и др.
Недифференцированная ДСТ — определяющий вариант ДСТ с клиническими проявлениями, не укладывающимися в структуру наследственных синдромов.
История
правитьНаучный и практический интерес к гипермобильности суставов возник ещё в конце XIX века, когда были описаны наследственные синдромы, в клинической картине которых гипермобильность суставов являлась одним из ведущих симптомов. Врачи различных специальностей регулярно видели пациентов, у которых имеет место структурное невоспалительное поражение (часто врождённое или проявляющееся в молодом возрасте) отдельных органов или систем.
Группа наследственных заболеваний соединительной ткани и скелета была впервые выделена американским генетиком Виктором Маккьюсиком в 1955 году. К тому времени она объединяла лишь некоторые нозологические формы: несовершенный остеогенез, синдром Марфана, синдром Элерса-Данло, эластическую псевдоксантому и гаргоилизм.
В 1967 году Кирк (J. H. Kirk), Анселл (B. M. Ansell) и Байватерс (E. G. Bywaters) предложили термин «гипермобильный синдром» для характеристики патологии у пациентов с гиперподвижными суставами и стойкими жалобами со стороны опорно-двигательного аппарата при отсутствии у них признаков какого-либо другого ревматического заболевания.[4] С этого времени началось систематическое изучение указанной патологии в рамках ревматологических синдромов. Термин «гипермобильный синдром» отражал феномен гипермобильности суставов, сочетающийся с дисфункцией опорно-двигательного аппарата (подвывихи, артралгии).[5]
Сегодня, благодаря достижениям генетики были описаны и классифицированы свыше 200 заболеваний соединительной ткани и скелета наследственного характера.[6]
-
 Гипермобильная кисть
Гипермобильная кисть -
 Гипермобильный пястно-фаланговый сустав I пальца
Гипермобильный пястно-фаланговый сустав I пальца -
 Гипермобильная кисть
Гипермобильная кисть -
 Гипермобильная кисть
Гипермобильная кисть -
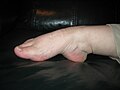 Гипермобильная лодыжка
Гипермобильная лодыжка -
 Гипермобильное плечо
Гипермобильное плечо -
 Гипермобильный палец
Гипермобильный палец -
 Гипермобильные лопатки
Гипермобильные лопатки -
 Гипермобильные локтевые суставы
Гипермобильные локтевые суставы








Терминология
правитьВ медицине уже более 100 лет известны дифференцированные соединительнотканные дисплазии: синдром Марфана, синдром Элерса-Данлоса, несовершенный остеогенез и др. (они включены в МКБ). Эти заболевания сравнительно редки и имеют чётко очерченные диагностические признаки. Однако существовала и группа пациентов, не удовлетворяющая критериям синдромной ДСТ. Очевидная генерализованность вовлечения в процесс соединительнотканных структур привела к широкому использованию обобщающих терминов: «соединительнотканная дисплазия», «врождённая соединительнотканная недостаточность», «недифференцированная наследственная коллагенопатия». В кардиологии распространено понятие соединительнотканных дисплазий сердца, МASS-фенотипа.
Отсутствие общепринятого определения не позволяло сравнивать и обобщать наблюдения разных авторов. Каждая новая публикация очередной раз констатировала «системность» вовлечения в процесс соединительнотканных структур при отдельном интересе авторов к одной из перечисленных в таблице нозологий.
В результате появился международный термин «гипермобильный синдром» (M35.7 в МКБ-10). Он не включил в себя дифференцированные формы соединительнотканной дисплазии. Достоинствами этого термина является выделение генерализованной гипермобильности суставов как наиболее характерного и легко определяемого клинического признака данной группы заболеваний, а отсутствие в определении слова «сустав» ориентирует врача на системные проявления синдрома.
В России чаще всего используют термин «дисплазия соединительной ткани», который включает в себя и синдромные и несиндромные формы. Иногда его также используют для обозначения только недифференцированной соединительнотканной дисплазии, общую группу наследственных коллагенопатий называя при этом «Наследственными нарушениями соединительной ткани».
Патогенез
правитьВ основе развития как ДСТ, так и ННСТ лежат мутации генов, ответственных за синтез/катаболизм структурных белков соединительной ткани или ферментов, участвующих в этих процессах, количественное изменение образования полноценных компонентов экстрацеллюлярного матрикса, нарушения фибриллогенеза (Яковлев В.М., Нечаева Г.И. 1994, Кадурина Т.И. 2009) [1, 7, 8]. Реализация генетических детерминант либо в наибольшей степени определяется внешними условиями, как в случае ДСТ, либо мало зависит от внешних условий, как в случае ННСТ [1-8]. В случаях ДСТ заболевание носит полигенно-мультифакториальный характер (заболевание с наследственной предрасположенностью), когда имеют место мутации большого количества генов, а случайная перекомбинация аллелей от отца и матери каждый раз приводит к формированию нового уникального генотипа [3-8]. Нутрициальные факторы, прежде всего, дефицит витаминов, макро- и микроэлементов, являются одной из основных причин, способствующих развитию ДСТ. Витамины группы В (В1, В2, В3, В6) нормализуют белковый обмен, витамин С и витамин Е поддерживают нормальный синтез коллагена, обладают антиоксидантной активностью. Макроэлементы (кальций, фосфор, магний) и микроэлементы (медь, цинк, селен, марганец, фтор, ванадий, кремний, бор) являются ко-факторами ферментов, активирующих синтез коллагена и минерализацию костной ткани. Микроэлементы также участвуют в водно-солевом и кислотно-щелочном обменах. Ионы калия, магния и цинка способствуют росту кости и поддерживают минеральную плотность костной ткани (Громова О.А., Торшин И.Ю., 2009) [4, 5, 9-11]. Все группы факторов вносят существенный вклад в развитие ДСТ.
Классификация
правитьКлассификация ДСТ – один из самых дискуссионных научных вопросов. Отсутствие единой, общепринятой классификации ДСТ отражает разногласие мнений исследователей по данной проблеме в целом. Учитывая, что классификация несёт важный «прикладной» смысл – используется как основа для формулировки диагноза и выбора тактики лечения, решение классификационных вопросов является очень важным с точки зрения клинической практики [3-8].
Выделяют дифференцированные (наследственные, синдромальные) и недифференцированные дисплазии соединительной ткани (ДСТ).
Суммарная частота наследственных ДСТ составляет доли процента, в то время как недифференцированные формы этой патологии имеют гораздо более широкое распространение, достигая в некоторых популяциях в среднем от 10 до 30%. Очевидно, что в своей практической деятельности врачи чаще сталкиваются именно с недифференцированными ДСТ. Однако наследственные ДСТ отличаются от недифференцированных форм в том отношении, что для многих из них в последние годы стали известны молекулярные основы этиологии и патогенеза. Поскольку спектры клинических проявлений этих двух групп ДСТ в значительной степени перекрываются, изучение относительно редких наследственных форм болезней соединительной ткани позволяет гораздо яснее понять причины возникновения и природу недифференцированных ДСТ, что имеет большое значение для выбора тактики ведения больных и предупреждения развития осложнений.
Под дифференцированной ДСТ понимают гетерогенную моногенную патологию, в основе которой лежат дефекты синтеза или катаболизма белков внеклеточного матрикса, либо белков, участвующих в морфогенезе соединительной ткани.
Для многих наследственных ДСТ характерна генетическая гетерогенность, обусловленная двумя особенностями наследования этих заболеваний - существованием аллельных серий и возможностью развития клинически сходных заболеваний при повреждении разных генов. Более половины наследственных ДСТ (57%) проявляется патологией скелета, более трети (36%) - нарушениями органа зрения, 40% из которых являются изолированными офтальмопатиями, около трети (27%) - изолированными или сочетанными черепно-лицевыми аномалиями, дефектами развития кистей и стоп. Практически каждая пятая наследственная ДСТ сопровождается нервно-мышечной (22%), диагностически значимой сердечно-сосудистой (18%) и кожной (18%) патологией. Реже дифференцированные ДСТ проявляются нарушениями слуха (14%), аномалиями зубов (13%), ногтей и/или волос (10%), желудочно-кишечного тракта (8%), бронхолёгочной (6%), мочевыделительной (4%) и половой (4%) систем.
Использование современных достижений молекулярной генетики позволяет выделить 8 групп наследственных ДСТ:
1. Наследственные коллагенопатии: несовершенный остеогенез, синдром Элерса-Данлоса, различные варианты хондродисплазий, офтальмопатий, нефропатий, аномалий суставов, органа зрения, миопатий, буллёзного эпидермолиза.
2. Наследственные фибриллинопатии: синдром Марфана; MASS-синдром; эктопия хрусталика с мягкими скелетными проявлениями марфаноидного типа без кардиоваскулярной патологии; синдром Марфана в сочетании с синдромом Шпринтцена-Гольдберга; марфаноидный скелетный синдром без кардиоваскулярных и глазных аномалий; синдром Вейля-Маркезани; контрактурная арахнодактилия, врождённая, или синдром Билса.
3. Наследственные эластинопатии: надклапанный стеноз аорты Эйзенберга; синдром Вильямса-Бурена; cutis laxa, врождённая, аутосомно-доминантная.
4. Наследственные фибулинопатии: cutis laxa, врождённая, аутосомно-доминантная; cutis laxa, врождённая, аутосомно-рецессивная; пятнистая дегенерация сетчатки, возрастзависимая, тип 1, тип 3; ячеистая дистрофия сетчатки.
5. Наследственные ламинопатии: мышечная дистрофия, врождённая, мерозиндефицитная, тип 1А; ларинго-онихо-кожный синдром, аутосомно-рецессивный; буллёзный эпидермолиз, линейный, летальный; буллёзный эпидермолиз, генерализованный, атрофический, доброкачественный; неонатальный cutis laxa с марфаноидным фенотипом; синдром микрокорнеа, врождённый нефроз Пирсона, перинатальная летальная форма.
6. Наследственные тромбоспондинопатии: псевдоахондроплазия, множественная эпифизарная дисплазия.
7. Наследственные протеогликанопатии: различные клинические варианты хондродисплазий, аномалий суставов, офтальмопатий, нефропатий и буллёзного эпидермолиза.
8. Наследственные ДСТ, обусловленные мутациями в генах фибробластных факторов роста, их рецепторов и антагонистов: различные формы краниосиностоза, ахондроплазии, хондродисплазии, брахидактилии, симфалангизма, синдромы множественного синостоза, анкилоза и склероостеоза.
Клинические проявления
правитьДСТ — это заболевание с очень разными клиническими проявлениями, от совсем лёгких до весьма серьёзных.
Клапанный синдром
правитьИзолированные и комбинированные пролапсы клапанов сердца (чаще всего пролапс митрального клапана — 70 %), миксоматозная дегенерация клапанов.
Торакодиафрагмальный синдром
правитьАстеническая форма грудной клетки, деформации грудной клетки (воронкообразная, килевидная), сколиоз, кифоз.
Сосудистый синдром
правитьПоражение артерий эластического типа: идиопатическое расширение стенки с формированием мешотчатой аневризмы; поражение артерий мышечного и смешанного типов: бифуркационно-гемодинамические аневризмы, долихоэктазии удлинённых и локальных расширений артерий, патологическая извитость вплоть до петлеобразования; поражение вен (патологическая извитость, варикозное расширение вен верхних и нижних конечностей, геморроидальных и др. вен); телеангиэктазии; эндотелиальная дисфункция.
Торакодиафрагмальное сердце
правитьАстенический, констриктивный, ложностенотический, псевдодилатационный варианты, торакодиафрагмальное лёгочное сердце.
Аритмический синдром
правитьЖелудочковая экстрасистолия различных градаций; многофокусная, мономорфная, реже полиморфная, монофокусная предсердная экстрасистолия; пароксизмальные тахиаритмии; миграция водителя ритма; атриовентрикулярные и внутрижелудочковые блокады; аномалии проведения импульса по дополнительным путям; синдром предвозбуждения желудочков; синдром удлинения интервала QT.
Бронхолёгочный синдром
правитьТрахеобронхиальная дискинезия, трахеобронхомаляция, трахеобронхомегалия, вентиляционные нарушения (обструктивные, рестриктивные, смешанные нарушения), спонтанный пневмоторакс.
Висцеральный синдром
правитьНефроптоз и дистопии почек, птозы органов желудочно-кишечного тракта, органов малого таза, дискинезии органов желудочно-кишечного тракта, дуоденогастральные и гастроэзофагеальные рефлюксы, несостоятельность сфинктеров, дивертикулы пищевода, грыжи пищеводного отверстия диафрагмы; птозы половых органов у женщин.
Синдром патологии органа зрения
правитьМиопия, астигматизм, гиперметропия, косоглазие, нистагм, отслоение сетчатки, вывих и подвывих хрусталика. Патология роговицы - кератоконус[7].
Геморрагические гематомезенхимальные дисплазии
правитьГемоглобинопатии, синдром Рандю–Ослера–Вебера, рецидивирующие геморрагические (наследственная дисфункция тромбоцитов, синдром Виллебранда, комбинированные варианты) и тромботические (гиперагрегация тромбоцитов, первичный антифосфолипидный синдром, гипергомоцистеинемия, резистентность фактора Vа к активированному протеину С) синдромы.
Синдром патологии стопы
правитьКосолапость, плоскостопие (продольное, поперечное), реже полая стопа.
Синдром гипермобильности суставов
правитьНестабильность суставов, вывихи и подвывихи суставов.
Вертеброгенный синдром
правитьмежпозвоночная грыжа, спондилолистез, сколиоз, кифоз, кифосколиоз.
Косметический синдром
правитьДиспластикозависимые дисморфии челюстно-лицевой области (аномалии прикуса, готическое нёбо, выраженные асимметрии лица); О- и Х-образные деформации конечностей; изменения кожных покровов (тонкая просвечивающаяся и легко ранимая кожа, повышенная растяжимость кожи, шов в виде «папиросной бумаги»).
Вторичные нарушения психики как следствие ДСТ
правитьСоматопсихические невротические расстройства, депрессия, ипохондрия (в т. ч. дисморфофобия), тревожно-фобические расстройства, нервная анорексия.
Пациенты с ДСТ формируют группу повышенного психологического риска, характеризующуюся сниженной субъективной оценкой собственных возможностей, уровнем претензий, эмоциональной устойчивости и работоспособности, повышенным уровнем тревожности, ранимостью, депрессивностью, конформизмом. Косметические дефекты в сочетании с астенией формируют психологические особенности этих больных: сниженное настроение, потеря ощущения удовольствия и интереса к деятельности, эмоциональная лабильность, пессимистическая оценка будущего, нередко с идеями самобичевания и суицидальными мыслями. Закономерным следствием психологического стресса является ограничение социальной активности, ухудшение качества жизни и значительное снижение социальной адаптации, наиболее актуальные в подростковом и молодом возрасте.[8]
Возможны также нарушения адаптации, аутизм.
Диагностика
правитьВедущее место при постановке диагноза любого наследственного заболевания принадлежит клинико-инструментальным, лабораторным, генеалогическим методам обследования больных и их семей. Клинические методы диагностики постоянно обновляются и совершенствуются. Без детального описания фенотипических проявлений заболевания у больных и членов их семей, составления на этой базе региональных и национальных регистров широкое внедрение современных молекулярно-генетических технологий в клиническую практику невозможно. Вместе с тем очевидно, что для многих наследственных ДСТ точный диагноз возможен только при молекулярной идентификации мутаций в соответствующих генах. Разработке методов молекулярной диагностики должна предшествовать работа по составлению баз данных семей с относительно однородными клиническими проявлениями заболевания. Уже при первых обращениях больных с тяжёлой, предположительно наследственной ДСТ необходимо направлять их в специализированные генетические центры для уточнения диагноза и выделения образцов ДНК.
Недифференцированные ДСТ диагностируются тогда, когда выявляемые у пациента фенотипические признаки не укладываются ни в одно из известных на сегодня наследственных заболеваний соединительной ткани. Недифференцированную ДСТ следует рассматривать как полиорганную и полисистемную патологию с прогредиентным течением, в основе которой лежит нарушение синтеза, распада или морфогенеза компонентов внеклеточного матрикса, возникающее у лиц с определённой генетической предрасположенностью в периоде раннего эмбриогенеза или постнатально под действием неблагоприятных факторов внешней среды. Следует признать, что на сегодня общепринятых критериев диагностики, единой терминологии недифференцированной ДСТ нет. Об этом красноречиво свидетельствует частота выявления этой патологии в популяции, которая, по данным разных авторов, колеблется от 8-9% и даже до 26 80%.
Недифференцированные ДСТ не входят в МКБ-10. На основании многолетнего практического опыта и результатов исследований считается, что постановка данного диагноза правомочна только при выявлении у пациента следующих признаков:
1) 6-8 и более клинико-инструментальных признаков ДСТ;
2) вовлечение в патологический процесс не менее 1-2 различных органов и систем;
3) лабораторное подтверждение факта нарушения обмена соединительной ткани;
4) выявление признаков семейного накопления проявлений ДСТ у родственников больного, обследованных по той же диагностической программе.
Если количество анализируемых клинических маркеров, выявляемых у пациента, меньше этого условно допустимого порога, то, более правильно констатировать факт накопления у него признаков ДСТ. Такой пациент требует динамического наблюдения, углублённого клинико-инструментального и лабораторного обследований с целью верификации диагноза.
В ряде случаев набор фенотипических признаков у пациентов напоминает ту или иную синдромальную патологию, которую следует расценивать как фенокопии наследственных ДСТ. Чаще всего недифференцированные ДСТ проявляются двумя известными фенотипами марфаноидным и элерсоподобным.
Для клиники недифференцированной ДСТ с марфаноидным фенотипом характерны астеническое телосложение, долихостеномелия, арахнодактилия, разной степени выраженности деформация грудной клетки, позвоночника, плоскостопие в сочетании с патологией клапанного аппарата сердца, реже с тенденцией к дилатации корня аорты и различными нарушениями органа зрения. Следует помнить, что данная формулировка диагноза правомочна лишь после тщательного исключения известных на сегодня наследственных фибриллинопатий.
При недифференцированной ДСТ с элерсоподобным фенотипом наблюдается сочетание признаков ДСТ с тенденцией к гиперрастяжимости кожи (обычно в пределах либо несколько превышающих 2,5 3 см) и разной степени суставов. Наиболее часто недифференцированные ДСТ с элерсоподобным фенотипом ассоциируются с патологией нервной системы. Данный тип ДСТ требует проведения дифференциального диагностики с синдромом Элерса -Данлоса, семейной гипермобильностью суставов, синдромом гипермобильности суставов, последствиями родовой травмы и др. Существенную помощь в диагностике недифференцированной ДСТ оказывают изучение особенностей течения раннего эмбрионального периода развития плода, а также клинико-генеалогический анализ с обязательным осмотром максимально возможного числа родственников пробанда.
См. также
правитьПримечания
править- ↑ Disease Ontology (англ.) — 2016.
- ↑ Monarch Disease Ontology release 2018-06-29 — 2018-06-29 — 2018.
- ↑ В. Б. Симоненко и др., Соединительнотканные дисплазия (Наследственные коллагенопатии), «Клиническая медицина», 2006, № 6, 62-68.
- ↑ J. H. Kirk, B. M. Ansell, E. G. Bywaters, The hypermobility syndrome, Ann. Rheum. Dis., 1967, 26, 425—427.
- ↑ А. Г. Беленький, Гипермобильный синдром — системное невоспалительное заболевание соединительной ткани, Кафедра ревматологии РМАПО, Москва. Дата обращения: 31 января 2019. Архивировано 12 февраля 2011 года.
- ↑ Гипермобильный синдром: клинические проявления, дифференциальный диагноз, подходы к терапии Н. Г. Правдюк, Н. А. Шостак , Кафедра факультетской терапии им. А. И. Нестерова, РГМУ, Москва. Дата обращения: 15 сентября 2009. Архивировано 23 октября 2009 года.
- ↑ М. М. Бикбов, В. К. Суркова, К. Х. Оганисян. Кератоконус как проявление соединительнотканных дисплазий. Офтальмология (31 марта 2015). Дата обращения: 8 августа 2020. Архивировано 23 сентября 2020 года.
- ↑ Дисплазия соединительной ткани: основные клинические синдромы, формулировка диагноза, лечение. Г. И. Нечаева, В. М. Яковлев, В. П. Конев, И. В. Друк, С. Л. Морозов — Лечащий врач. Дата обращения: 18 июля 2009. Архивировано 15 июля 2009 года.
Ссылки
править- Группа ВКонтакте людей, страдающих синдромом Марфана
- «Дисплазия соединительной ткани» — Медицинский информационный сайт (Омский государственный медицинский университет)
- «Принципы реабилитации больных с дисплазией соединительной ткани» — Медицинский научно-практический портал «Лечащий врач», № 04/10 Коллоквиум